

Scientific Reports volume 14, Article number: 21439 (2024)
Abstract
In this study, we successfully established a novel gallbladder cancer cell line, designated as GBC-X1, derived from a primary tumor of a gallbladder cancer patient. By comprehensively analyzing the cell line’s phenotype, molecular characteristics, biomarkers, and histological characteristics, we confirmed that GBC-X1 serves as a valuable model for investigating the pathogenesis of gallbladder cancer and developing therapeutic agents. GBC-X1 has been continuously cultured for one year, with over 60 stable passages. Morphologically, GBC-X1 exhibits typical features of epithelial tumors. The population doubling time of GBC-X1 is 32 h. STR analysis validated a high consistency between GBC-X1 and the patient’s primary tumor. Karyotype analysis revealed an abnormal hypertetraploid karyotype for GBC-X1, characterized by representative karyotypes of 98, XXXX del (4) p (12) del (5) p (21) der (10). Under suspension culture conditions, GBC-X1 efficiently forms tumor balls, while subcutaneous inoculation of GBC-X1 cells into NXG mice leads to xenograft formation with a rate of 80%. Drug sensitivity testing demonstrated that GBC-X1 is resistant to oxaliplatin and sensitive to 5-FU, gemcitabine, and paclitaxel. Immunohistochemistry revealed positive expression of CK7, CK19, E-cadherin, MMP-2, CD44, SOX2, and TP53 in GBC-X1 cells, weak positive expression of Vimentin, and a Ki67 positive rate of 35%. Our research highlights GBC-X1 as a novel gallbladder cancer cell line and emphasizes its potential as an effective experimental model for investigating the pathogenesis of gallbladder cancer and drug development.
Similar content being viewed by others


Introduction
Gallbladder cancer commonly originates from the mucosal epithelium of the gallbladder and represents the most prevalent and highly prognostically unfavorable malignant tumor in the biliary system1,2. In 2020, there were approximately 19.3 million newly diagnosed cancer cases worldwide, resulting in nearly 10 million cancer-related deaths. Among these cases, gallbladder cancer accounted for 0.6% of newly diagnosed cancer patients and 0.9% of cancer-related deaths3. Gallbladder cancer has obvious geographical distribution characteristics, among which Bolivia in South America, Chile, northern India, Eastern Europe, and some Southeast Asian countries have higher incidence rate; The incidence rate is the lowest in European origin regions such as the United States, Australia, Canada, the United Kingdom and New Zealand4,5. Gallbladder cancer is one of the few tumors showing global gender differences. The incidence rate of women is three to six times higher than that of men6. The overall incidence rate in China is about 3.82/100,000, of which the incidence rate in Shanghai can reach 7.8/100,000. In recent years, the incidence rate in China has been rising, causing more and more attention7,8. Radical surgery remains the sole approach to achieving long-term survival for gallbladder cancer patients. However, this disease exhibits covert initial onset, marked malignancy, pronounced invasiveness, and a propensity for metastasis and dissemination. Early diagnosis is only possible for a meager 10–20% of patients9,10, with the majority being diagnosed at advanced stages, thus missing the opportunity for surgical intervention. The 5-year survival rate for patients with advanced gallbladder cancer ranges from a mere 5–15% 4,11.
In 1980, Koyama established the world’s first gallbladder cancer cell line, G-415. Since then, researchers from Japan and Korea have subsequently established multiple gallbladder cancer cell lines11,12. These cell lines closely mirror the characteristics of primary tumors, rendering the insights derived from their study more representative of the actual in vivo conditions13. Given the variation in tumor etiology, tumor heterogeneity, and genetic diversity influenced by race14,15,16,17, the establishment of a diverse range of gallbladder cancer cell lines assumes significant importance in facilitating basic research and drug development pertaining to this disease.
In this study, we successfully established a stable gallbladder cancer cell line, designated as GBC-X1, using surgically resected tumor tissue obtained from a gallbladder cancer patient. GBC-X1 represents a valuable experimental model for gallbladder cancer research.
Materials and methods
Source of human tissue
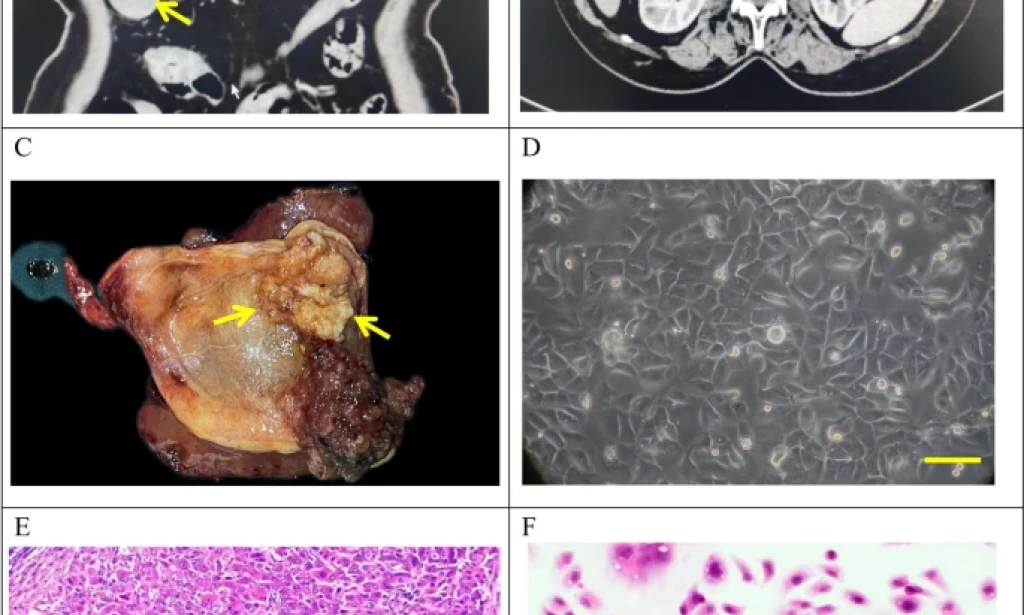
The tumor tissue was obtained from a 72-year-old female patient who underwent radical cholecystectomy at Lanzhou University Second Hospital on May 9, 2022. The patient presented with a gallbladder tumor, had no history of smoking and drinking, and had a past medical history of hepatitis B infection. Preoperative laboratory tests showed CEA levels of 9.9 ng/ml (reference range: 0-5.2 ng/ml), AFP levels of 1.2 U/ml (reference range: 0-5.8 U/ml), and CA19-9 levels of 49.6 U/ml (reference range: 0-35U/ml) (Table 1). Preoperative CT findings confirmed the presence of gallbladder cancer (Fig. 1A-B).
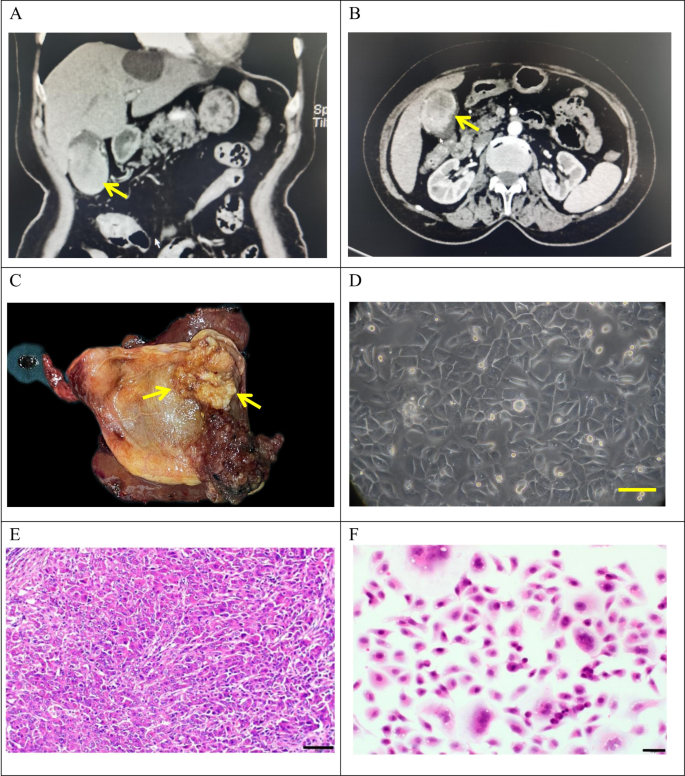
Clinical and pathological profle of GBC-X1.A and B: The patient’s preoperative CT scan results showed a huge mass at the bottom of the gallbladder (arrow). C: On the gross view of the postoperative specimen, a cauliflower-like mass can be seen in the gallbladder (arrow). D: Morphology of GBC-X1 cells under light microscopy ( scale bar = 50 μm). E: The patient’s primary tumor showed moderately to poorly differentiated. The tumor cells displayed irregular arrangements in glandular, tubular, cord-like, and nest-like patterns (scale bar = 50 μm). F: GBC-X1 cells were varying shapes, primarily short fusiform, characterized by enlarged nuclei, prominent nucleoli, limited cytoplasm, and the presence of multinucleated and megakaryocytic cells (scale bar = 50 μm).
A 4 × 4 cm neoplasm was observed in the gallbladder (Fig. 1C). Tissues were collected from the primary lesion for primary culture and subculture.
This study was approved by the Medical Ethics Committee of Lanzhou University Second Hospital (2023 A-381), and the patient provided an informed consent form.
Drugs
Gemcitabine was obtained from Jiangsu Haosen Pharmaceutical Group Co., Ltd., oxaliplatin from Jiangsu Hengrui Pharmaceutical Co., Ltd., 5-FU from Tianjin Jinyao Pharmaceutical Co., Ltd., and paclitaxel from Jiangsu Aosaikang Pharmaceutical Co., Ltd.
NXG mice
Two batches of NXG mice (2 and 3 mice), aged 5–6 weeks, weighing 11 g and 17 g, were used as experimental animals. The mice were purchased from Changzhou Cavens Experimental Animal Co., Ltd. and housed in the SPF level laboratory of the Animal Experimental Center at Lanzhou University. The animal protocol was designed to minimize pain or discomfort to the animals. The animals were acclimatized to laboratory conditions (23℃, 12 h/12 h light/dark, 50% humidity, ad libitum access to food and water) for 2 wk prior to experimentation. The animals were fed an autoclaved rodent diet ad libitum. The mice bedding, feed and water were replaced every 2 days. All procedures followed the institutional and national guidelines for the care and use of laboratory animals. Animal health and behavior were monitored every day for 4 weeks. When the maximum diameter of the Xenograft tumor approaches 1.5 cm or when the Xenograft tumor grows for one month, the mice were killed. All animals were euthanized by barbiturate overdose (intravenous injection, 150 mg/kg pentobarbital sodium) for tissue collection. Death was confirmed by loss of heartbeat, breathing and pupil response.
Animal experiments were reviewed and approved by the Medical Animal Experiment Ethics Committee of Lanzhou University Second Hospital (D2023-318). The all experiments were conducted in accordance with the ARRIVE guidelines and the Guidelines for the Care and Use of Laboratory Animals of China.
The following methods were similar or identical to those employed in previous studies18.
Primary culture, cell purification, and cell line establishment
The tumor tissue was rinsed with sterile PBS (catalog no.10010023, Gibco, USA) 3–5 times, minced as finely as possible, and then digested with type II collagenase (catalog no.17101015, Gibco, USA) and dispase (catalog no.17105041, Invitrogen, USA) in a shaking incubator at 37 ℃. Once the tissue blocks were partially digested, the supernatant was collected, filtered through a 100-mesh filter, and centrifuged at 300 g for 3 min. The supernatant was discarded, and the cell pellet was resuspended in PBS, followed by centrifugation at 300 g for 3 min. The entire culture medium (RPMI-1640(catalog no.c3010-0500, BI, Israel) + 10% FBS (catalog no.04-002-1 A, BI, Israel) + 1% penicillin-streptomycin (catalog no.03-031-1B, BI, Israel)) was added to the pellet and thoroughly mixed. The cell suspension was then evenly seeded into a six-well plate (catalog no.703001, NEST, China). The culture medium was changed after 48 h. Mechanical scraping was performed to remove any fibroblast contamination during the primary culture process. When the cells reached 80% confluency, they were trypsinized and passaged. The cell growth status was regularly monitored under an optical microscope. Starting from the third generation, cells were passaged at a 1:2 ratio and cryopreserved using a serum-free rapid cell cryopreservation solution (catalog no.MF699-01, Mei5 Biotechnology Co.,Ltd, China).
Cell growth curve
GBC-X1 cells (P25) in the logarithmic growth phase were trypsinized and adjusted to a cell density of 6 × 104/ml. Then 0.1 ml of the cell suspension was inoculated into each well of a 96-well plate. CCK-8 (catalog no.ck04, Dojindo, Japan) reagent was added simultaneously and incubated for 2 h for four consecutive days. The UV absorbance value at a wavelength of 450 nm was measured using an enzyme-linked immunosorbent assay reader, and a standard curve was plotted. Population doubling time were computed with several time points, using the “cell calculator++”tool [V. Roth MD, Doubling Time Calculator (2006), https://doubling-time.com/compute_more.php].
Short tandem repeat analysis
GBC-X1 cells (P15) in the logarithmic growth phase were collected after trypsin digestion (catalog no.03-050-1BCS, BI, Israel). STR analysis was performed by Suzhou Genetic Testing Biotechnology Company to establish the correlation between the cells and the primary tumor tissue.
Karyotype analysis
GBC-X1 cells (P42) in the logarithmic growth phase were treated with 0.25 µg/mL colchicine for 6 h at 37 ℃. Metaphase cells were collected, and fixed with methanol acetic acid (3:1). After trypsin digestion, the specimens were stained with Giemsa staining solution. The specimens were analyzed under a microscope. Select well dispersed and moderately stained mitotic phases for karyotype analysis.
Tumor sphere formation assay
Logarithmically growing cells were collected, and 1.5 × 105 cells were inoculated per well into a 6-well plate (Ultra-Low attachment Plate)(catalog no.3471, Corning, USA) after trypsin digestion. The cells were cultured in serum-free RPMI-1640 medium, and tumor sphere formation was assessed on days 7, 10, and 14 after seeding.
Scanning electron microscope
Cells from 30 passages in the logarithmic growth phase were used for the experiments. Cell slides were washed with physiological saline and quickly immersed in 4% glutaraldehyde (product of SPI-CHEM, USA) fixative solution. After rinsing three times with phosphate buffer, the cells were dehydrated using a stepwise gradient of tert-butanol (50%, 70%, 80%, 90%, 100%) for 5 min each. The dried samples were then placed in a JEOL JFD-320 cold dryer, and once the temperature reached room temperature, they were coated with a conductive paste using a JEOL JFCC-160 ion sputterer. The samples were then observed and photographed using a HITACHI Regulus 8100 scanning electron microscope.
Transmission electron microscope
Logarithmically growing GBC-X1 cells (P33) were collected by centrifugation. The cells were fixed in a fixative solution of 2% Paraformaldehyde − 2.5% Glutaraldehyde, followed by flushing with 0.1 mol/L dimethyl Sodium arsenate buffer (pH = 7.4). Subsequently, the cells were further fixed with 1% osmium tetroxide, washed with double-distilled water, dehydrated with a gradient of ethanol to propylene oxide, and embedded in SPI812 resin. Semi-thin sections of 1 µM using the Leica EMUC7 slicing mechanism. After observation and positioning under a light microscope with azure methylene blue staining, 60 nm ultra-thin sections were prepared using Leica EMUC7 slicing mechanism. These sections were stained with uranyl acetate and lead citrate and subjected to observation and image analysis using HITACHI H-7650 Transmission electron microscopy.
Drug sensitivity experiment
Logarithmically growing GBC-X1 cells (P45) were trypsinized and prepared as a single-cell suspension. A total of 10,000 cells per 100 µl were inoculated into each well of a 96-well plate, with six wells repeated in each group. After cells adhered to the wall, the experimental group was treated with different concentrations of anti-tumor drugs, while the control group was treated with corresponding drug solubilizers. After 72 h of drug action, the complete medium was replaced with 100 µl serum-free medium containing 10% (v/v) CCK8. After a 2-hour incubation, the optical density (OD) value at 450 nm was measured.
Transwell chamber migration assay
Cultivated cells in serum-free medium for 1 day in advance. GBC-X1 cells (P40) were collected after trypsin digestion, adjusted the cell concentration to 7.5 × 105/mL, and added 200ul of cell suspension to the upper chamber of Transwell. Added 700 ul of complete culture medium containing 15% FBS to the lower chamber of Transwell. Cultivated them for 48 h. Discarded the culture medium from the upper and lower chambers, gently washed the upper and lower chambers 1–2 times with 1 ml PBS, added 1 ml of 4% paraformaldehyde, fixed for 15 min, and then discarded. Added 1.5 ml of 0.1% crystal violet staining solution to the Transwell chamber, stained for 30 min, and discarded. Washed the Transwell upper chamber three times in ultrapure water, washed the bottom of the upper chamber membrane with a wet cotton ball, and then washed again once. Placed the Transwell upper chamber on a glass slide and observed and taked photos under a microscope.
Transwell chamber invasion assay
Cultivated cells (P40) in serum-free medium for 1 day. Diluted the Matrigel with serum-free medium at a ratio of 1:8 under 4℃ conditions. Took 50 ul of matrigel and added it to the Transwell upper chamber, evenly covering the polycarbonate film. Placed the Transwell upper chamber in a 37℃ cell culture incubator for 30 min, then discarded any excess liquid in the upper chamber. Collected the cells with trypsin, and adjusted the cell concentration to 7.5 × 105/mL, and added 200ul of cell suspension to the upper chamber of Transwell. Added 700 ul of complete culture medium containing 15% FBS to the lower chamber of Transwell. Cultivated them for 48 h. Discarded the culture medium from the upper and lower chambers, gently washed the upper and lower chambers 1–2 times with 1 ml PBS, added 1 ml of 4% paraformaldehyde, fixed for 15 min, and then discarded. Added 1.5 ml of 0.1% crystal violet staining solution to the Transwell chamber, stained for 30 min, and discarded. Washed the Transwell upper chamber three times in ultrapure water, washed the bottom of the upper chamber membrane with a wet cotton ball. Placed the Transwell upper chamber on a glass slide and observed and took photos under a microscope.
Xenograft formation experiment
Logarithmically growing cells (P40 and P45) were trypsinized and adjusted to a cell density of 1 × 107/ml. A total of 0.1 ml of cell suspension was inoculated on the right shoulder of five NXG mice. Tumor growth of the nude mice was observed and recorded the next day. After 4 weeks, the mice were euthanized, and the xenograft growth was observed upon dissection.
Immunohistochemical staining
Cells from the 42nd passage were digested and inoculated onto sterile slides for growth. After 48 h, the slides were washed with PBS, fixed with 4% paraformaldehyde for 15 min, air-dried, and treated with 0.5% Triton X-100 for 20 min. Paraffin sections of xenograft and primary tumors were prepared and baked overnight at 60 ℃. Dewaxing, gradient alcohol hydration, and antigen retrieval were performed using Dako’s Autostainer Link 48 instrument. The activity of peroxidase was blocked by incubating the slides with 3% hydrogen peroxide solution at 37 °C for 15 min. Then, 100 µL of normal goat serum was added dropwise and sealed at 37 ℃ for 15 min. The primary antibodies (CK7(catalog no. kit-0021, MXB, China), CK19(catalog no.kit-0030, MXB, China), Ki67(catalog no.RMA-0731, MXB, China), E-cadherin(catalog no.MAB-0738, MXB, China), Vimentin(catalog no. MAB-0735, MXB, China), MMP-2(catalog no. GB114959-50, Servicebio, China), CD44(catalog no. GB113500-50, Servicebio, China), SOX2(catalog no. GB11249-50, Servicebio, China), TP53(catalog no. GB111740-50, Servicebio, China) ) were incubated at 37 °C for 1 h. Color development was performed using the DAB staining solution kit (Dako), followed by rinsing with running water for 5 min. The slides were restrained with hematoxylin, dehydrated, and made transparent in xylene. Finally, the slides were sealed with neutral resin and observed under a microscope.
Statistical analysis
All statistical analyses were conducted using SPSS 26.0 software. The data were presented as mean ± SD. Student’s t-tests and ANOVA were used for group comparisons. A P-value of < 0.05 was considered statistically significant.

You must be logged in to post a comment.